 A Robot’s Sigh
A Robot’s Sigh
lammpstrj-parserの使い方
はじめに
LAMMPSという分子動力学法のパッケージがあります。実行すると、lammpstrjというダンプファイルを吐きます。このダンプファイルに原子の座標や速度情報が入っているので、それをいろいろ解析するのですが、Pythonで書くと大規模な時に時間がかかり過ぎたり、C++で毎回書くのは面倒だったりします。なので、lammpstrjファイルをパースするC++ライブラリを作りました。
これはC++のシングルヘッダーライブラリです。lammpstrj::for_each_frameに
lammpstrj::for_each_frame(filename, callback_function);
のようにlammpstrjファイル名とコールバック関数を登録すると、フレームごとにコールバック関数に原子の情報が渡されて呼ばれます。なのでその中でいろいろすればOKです。
以下では、その使い方を簡単に紹介します。
完成するサンプルは
kaityo256/lammpstrj-parser-sample
に置いておきます。
クローンとコンパイル
シングルヘッダーライブラリですが、git submoduleとして使うのが楽です。なので、まずそれを使うためのリポジトリを作りましょう。ディレクトリ名はなんでも良いですが、lammpstrj-parser-sampleとしておきます。
mkdir lammpstrj-parser-sample
cd lammpstrj-parser-sample
git init .
リポジトリを作ったら、lammpstrj-parserをsubmoduleとして登録します。こちらも場所はどこでも良いですが、external/lammpstrj-parserに追加しましょう。
git submodule add https://github.com/kaityo256/lammpstrj-parser external/lammpstrj-parser
次にmain.cppを作成します。
#include <lammpstrj/lammpstrj.hpp>
int main() {
}
さらに、Makefileを作ります。ライブラリへのパスexternal/lammpstrj-parser/includeをコンパイラに教える必要があります。
CXX = g++
CXXFLAGS = -std=c++14 -O2 -Iexternal/lammpstrj-parser/include
all: lammpstrj-sample
lammpstrj-sample: main.cpp
$(CXX) $(CXXFLAGS) main.cpp -o $@
.PHONY: clean
clean:
rm -f lammpstrj-sample
この状態でビルドできることを確認しましょう。
make
エラーなしにビルドできたら、ライブラリのインストールに成功しています。
.gitignoreを追加してからコミットしておきましょう。
lammpstrj-sample
*.lammpstrj
log.lammps
*.dat
*.png
*.vtk
*.gif
コミットする前はこんな状態になっているはずです。
$ git status -sA .gitmodules
A external/lammpstrj-parser
?? .gitignore
?? Makefile
?? main.cpp
コミットしましょう。
git add .
git commit -am ":tada: initial commit"
LAMMPSの準備と実行
インプットファイルの準備
同じディレクトリにLAMMPSのインプットファイルを作りましょう。二種類の原子の相分離シミュレーションをしてみます。
こんな内容のtest.inputファイルを作ります。
units lj
atom_style atomic
boundary p p p
timestep 0.001
variable rho equal 0.5
variable L equal 20.0
lattice fcc ${rho}
region box block 0 ${L} 0 ${L} 0 ${L}
create_box 2 box
create_atoms 1 box
set type 1 type/fraction 2 0.5 98765
mass 1 1.0
mass 2 1.0
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
pair_coeff 1 2 1.0 1.0 1.12246
pair_coeff 2 2 1.0 1.0 2.5
velocity all create 1.0 12345 mom yes rot yes dist gaussian
fix 1 all nvt temp 0.7 0.7 1.0
dump 1 all custom 500 test.lammpstrj id type x y z vx vy vz
thermo 500
run 5000
以下、内容の説明です。まず、密度0.5のFCCで20x20x20のブロックで原子を配置します。
variable rho equal 0.5
variable L equal 20.0
lattice fcc ${rho}
region box block 0 ${L} 0 ${L} 0 ${L}
create_box 2 box
create_atoms 1 box
そして、そのうち半分の原子タイプを2に変更します。
set type 1 type/fraction 2 0.5 12345
次に、二種類の原子の質量や相互作用を定義します。
mass 1 1.0
mass 2 1.0
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
pair_coeff 1 2 1.0 1.0 1.12246
pair_coeff 2 2 1.0 1.0 2.5
同種原子(1-1や2-2)間はカットオフ2.5おLennard-Jonesポテンシャル、異種原子(1-2)間は、カットオフ1.12245のLennard-Jonesポテンシャルにしています。1.12245は$2^(1/6)$で、ここをカットオフにすると、斥力相互作用だけが残ります。これにより、同種原子間には引力が、異種原子間には斥力のみが働くことになります。
あとは適当な初期速度を与え、温度を0.7に制御します。さらに、500ステップごとにtest.lammpstrjにダンプを吐きます。デフォルトでは座標しか出力されないため、カスタムで速度も出します。
velocity all create 1.0 12345 mom yes rot yes dist gaussian
fix 1 all nvt temp 0.7 0.7 1.0
dump 1 all custom 500 test.lammpstrj id type x y z vx vy vz
最後に500ステップに一度熱力学量を出力するようにして、まずは短めに5000ステップ実行しましょう。
thermo 500
run 5000
LAMMPSの実行
先ほどのインプットファイルを食わせて実行してみましょう。
$ lmp_serial -i test.input
LAMMPS (20 Nov 2019)
Lattice spacing in x,y,z = 2 2 2
Created orthogonal box = (0 0 0) to (40 40 40)
1 by 1 by 1 MPI processor grid
Created 32000 atoms
(snip)
Step Temp E_pair E_mol TotEng Press
0 1 -1.5157574 0 -0.01580429 -0.82634209
500 1.0508477 -1.9127298 0 -0.3365075 0.93704824
1000 1.0606635 -1.9990148 0 -0.40806932 0.87624785
1500 1.0504821 -2.0771359 0 -0.50146201 0.82737376
2000 1.0325874 -2.1528941 0 -0.6040615 0.77292665
2500 1.0051272 -2.2201976 0 -0.71255391 0.71308458
3000 0.98362265 -2.2979528 0 -0.82256491 0.64688919
3500 0.96133378 -2.3729887 0 -0.93103311 0.58650952
4000 0.93291535 -2.4358995 0 -1.0365702 0.54039314
4500 0.91849522 -2.5155906 0 -1.1378909 0.47440337
5000 0.88856984 -2.5708378 0 -1.2380246 0.43675265
(snip)
後のためにシステムサイズや原子数、途中の温度の時間発展だけ確認しておきます。 システムサイズが40x40x40で、原子数が32000であることがわかります。
温度の計算
システム情報の取得
では、lammptrj-parserを使ってみましょう。main.cppのmain関数を以下のように修正します。
#include <cstdio>
#include <lammpstrj/lammpstrj.hpp>
#include <string>
int main() {
const auto filename = "test.lammpstrj";
auto si = lammpstrj::read_info(filename);
printf("(LX, LY, LZ) = (%f, %f, %f)\n", si->LX, si->LY, si->LZ);
printf("N = %d\n", si->atoms);
}
lammpstrj::read_info関数にファイルを指定すると、lammpstrj::SystemInfo構造体が返ってきます。
auto si = lammpstrj::read_info(filename);
si->LXなどにシステムサイズに、si->atomsに原子数が入っています。
ビルドして実行してみましょう。
$ make
g++ -std=c++14 -O2 -Iexternal/lammpstrj-parser/include main.cpp -o lammpstrj-sample
$ ./lammpstrj-sample
(LX, LY, LZ) = (40.000000, 40.000000, 40.000000)
N = 32000
先ほど確認したように、システムサイズが40x40x40、原子数が32000であり、正しく情報が取れていることがわかります。
温度の取得
次に、コールバック関数を作ります。温度を計算するための関数calc_temperatureを作りましょう。
void calc_temperature(const std::unique_ptr<lammpstrj::SystemInfo> &si, const std::vector<lammpstrj::Atom> &atoms) {
static int frame_ = 0;
double e = 0.0;
for (auto &a : atoms) {
e += a.vx * a.vx + a.vy * a.vy + a.vz * a.vz;
}
e /= static_cast<double>(si->atoms);
e /= 3.0;
printf("%d %f\n", frame_ * 500, e);
frame_++;
}
引数はconst std::unique_ptr<lammpstrj::SystemInfo> &si, const std::vector<lammpstrj::Atom> &atomsです。システム情報の構造体lammpstrj::SystemInfoと、原子の情報const std::vector<lammpstrj::Atom> &atomsが渡されるので、速度の自乗和から音頭を計算します。
main関数の最後にlammpstrj::for_each_framにファイル名とcalc_temperature関数を渡して実行するようにします。
int main() {
const auto filename = "test.lammpstrj";
auto si = lammpstrj::read_info(filename);
printf("(LX, LY, LZ) = (%f, %f, %f)\n", si->LX, si->LY, si->LZ);
printf("N = %d\n", si->atoms);
lammpstrj::for_each_frame(filename, calc_temperature); // ここを追加
}
ビルド、実行してみましょう。
$ make
g++ -std=c++14 -O2 -Iexternal/lammpstrj-parser/include main.cpp -o lammpstrj-sample
$ ./lammpstrj-sample
(LX, LY, LZ) = (40.000000, 40.000000, 40.000000)
N = 32000
0 0.999969
500 1.050815
1000 1.060630
1500 1.050449
2000 1.032555
2500 1.005096
3000 0.983592
3500 0.961304
4000 0.932886
4500 0.918467
5000 0.888542
先ほどの出力と比較したら1フレームだけずれているせいで値が少しずれていますが、正しく温度が取れていることがわかります。
コールバック関数は生の関数にしても良いですが、後々のためにクラスにまとめておきましょう。TemperatureCalculatorを作って、const std::string filename_;、int frame_をメンバ変数に、calc_temperatureをメンバ関数にします。
class TemperatureCalculator{
private:
const std::string filename_;
int frame_;
void calc_temperature(const std::unique_ptr<lammpstrj::SystemInfo> &si, const std::vector<lammpstrj::Atom> &atoms) {
static int frame_ = 0;
double e = 0.0;
for (auto &a : atoms) {
e += a.vx * a.vx + a.vy * a.vy + a.vz * a.vz;
}
e /= static_cast<double>(si->atoms);
e /= 3.0;
printf("%d %f\n", frame_ * 500, e);
frame_++;
}
public:
TemperatureCalculator(const std::string filename): filename_(filename){
frame_ = 0;
}
void analyze(void){
auto callback_function = [this](const std::unique_ptr<lammpstrj::SystemInfo> &si, const std::vector<lammpstrj::Atom> &atoms){calc_temperature(si, atoms);};
lammpstrj::for_each_frame(filename_, callback_function);
}
};
コンストラクタでfilename_を受け取り、frame_を0に初期化します。
クラスのメンバ関数はそのままではコールバック関数として渡せないため、少し工夫が必要です。いくつか方法がありますが、一番簡単なのはラムダ式を使うことです。
void analyze(void){
auto callback_function = [this](const std::unique_ptr<lammpstrj::SystemInfo> &si, const std::vector<lammpstrj::Atom> &atoms){calc_temperature(si, atoms);};
lammpstrj::for_each_frame(filename_, callback_function);
}
callback_functionというラムダ式を作り、ラムダ式の中でメンバ関数を呼ぶようにします。それをlammpstrj::for_each_frameに渡します。
このanalyze関数をmain関数が呼べばOKです。
int main() {
const auto filename = "test.lammpstrj";
auto si = lammpstrj::read_info(filename);
printf("(LX, LY, LZ) = (%f, %f, %f)\n", si->LX, si->LY, si->LZ);
printf("N = %d\n", si->atoms);
//lammpstrj::for_each_frame(filename, calc_temperature);
// 以下を追加
TemperatureCalculator tc(filename);
tc.analyze();
}
ビルド、実行すると先程と同じ結果が得られます。
ファイルへの出力
いま、温度を標準出力に吐いていましたが、出力するものが増えてくるとファイルに吐いたほうが便利になります。なので、温度をtemperature.datに出力するようにしましょう。コールバック関数で毎回ファイルに追記する必要があるため、クラスのコンストラクタでファイルを開いておく必要があります。
まず、メンバ変数としてstd::ofstream ofs_;を追加します。
class TemperatureCalculator {
private:
const std::string filename_;
int frame_;
std::ofstream ofs_; // ここを追加。
そして、コンストラクタでファイルを開きます。
TemperatureCalculator(const std::string filename) : filename_(filename) {
frame_ = 0;
ofs_.open("temperature.dat"); //ここを追加
}
あとは、標準出力に出していたのをofs_に変えるだけです。
void calc_temperature(const std::unique_ptr<lammpstrj::SystemInfo> &si, const std::vector<lammpstrj::Atom> &atoms) {
static int frame_ = 0;
double e = 0.0;
for (auto &a : atoms) {
e += a.vx * a.vx + a.vy * a.vy + a.vz * a.vz;
}
e /= static_cast<double>(si->atoms);
e /= 3.0;
// printf("%d %f\n", frame_ * 500, e);
ofs_ << frame_ * 500 << " " << e << std::endl; // ここを追加
std::cerr << frame_ * 500 << std::endl; //ここも
frame_++;
}
ついでに標準エラーに進捗を吐いておくと便利です。これで実行すると進捗としてフレーム数が表示されつつ、温度がtemperature.datに出力されるようになります。
$ ./lammpstrj-sample
(LX, LY, LZ) = (40.000000, 40.000000, 40.000000)
N = 32000
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
$ cat temperature.dat
0 0.999969
500 1.05081
1000 1.06063
1500 1.05045
2000 1.03256
2500 1.0051
3000 0.983592
3500 0.961304
4000 0.932886
4500 0.918467
5000 0.888542
密度の取得とVTKファイルの出力
メッシュサイズの計算
せっかく相分離シミュレーションをしているので、その様子を可視化してみましょう。シミュレーションボックスを小さなセルに分割し、各セルで局所密度を計算、その内容をVTKファイルとして出力します。とりあえず必要なメンバ変数を定義し、メッシュサイズを計算するコンストラクタを作ります。
class LocalDensityCalculator {
private:
const std::string filename_;
int frame_;
double mesh_size_;
int nx_, ny_, nz_, total_cells_;
double mx_, my_, mz_;
public:
LocalDensityCalculator(const std::string filename, const double mesh_size): filename_(filename), mesh_size_(mesh_size){
frame_ = 0;
auto si = lammpstrj::read_info(filename);
// セル数(切り上げ)
nx_ = static_cast<int>(std::ceil(si->LX / mesh_size_));
ny_ = static_cast<int>(std::ceil(si->LY / mesh_size_));
nz_ = static_cast<int>(std::ceil(si->LZ / mesh_size_));
total_cells_ = nx_ * ny_ * nz_;
// 実際のセルサイズ
mx_ = si->LX / static_cast<double>(nx_);
my_ = si->LY / static_cast<double>(ny_);
mz_ = si->LZ / static_cast<double>(nz_);
std::cerr << "Requested mesh size: " << mesh_size_ << std::endl;
std::cerr << "Actual cell size (mx, my, mz): "
<< mx_ << ", " << my_ << ", " << mz_ << std::endl;
std::cerr << "Number of cells (nx, ny, nz): "
<< nx_ << ", " << ny_ << ", " << nz_ << std::endl;
}
};
コンストラクタでlammpstrjファイルのファイル名と、メッシュサイズを受け取ります。そして、実際のメッシュサイズを出力します。これは、メッシュサイズでシステムサイズが割り切れない場合があるためです。
main関数から読んでみましょう。
int main() {
const auto filename = "test.lammpstrj";
auto si = lammpstrj::read_info(filename);
printf("(LX, LY, LZ) = (%f, %f, %f)\n", si->LX, si->LY, si->LZ);
printf("N = %d\n", si->atoms);
//lammpstrj::for_each_frame(filename, calc_temperature);
//TemperatureCalculator tc(filename);
//tc.analyze();
LocalDensityCalculator lc(filename, 2.0); // ここを追加。
}
ビルド、実行すると以下のような結果になります。
$ make
g++ -std=c++14 -O2 -Iexternal/lammpstrj-parser/include main.cpp -o lammpstrj-sample
$ ./lammpstrj-sample
(LX, LY, LZ) = (40.000000, 40.000000, 40.000000)
N = 32000
Requested mesh size: 2
Actual cell size (mx, my, mz): 2, 2, 2
Number of cells (nx, ny, nz): 20, 20, 20
システムサイズが40x40x40で、メッシュサイズが2.0を要求したので、この場合は割り切れて、実際のメッシュサイズが2x2x2、セルの数が20x20x20になりました。
では、メッシュサイズとして3.0を要求するとどうなるでしょうか。
LocalDensityCalculator lc(filename, 3.0); // メッシュサイズを3.0にしてみる。
実行結果は以下のようになります。
$ ./lammpstrj-sample
(LX, LY, LZ) = (40.000000, 40.000000, 40.000000)
N = 32000
Requested mesh size: 3
Actual cell size (mx, my, mz): 2.85714, 2.85714, 2.85714
Number of cells (nx, ny, nz): 14, 14, 14
メッシュサイズとして、3.0に近い2.85714が選ばれ、メッシュの数は14x14x14になりました。このように、メッシュの数が整数になるように、要求メッシュサイズに最も近い値が選ばれます。
動作確認後、メッシュサイズを2.0に戻しておきましょう。
VTKファイルの出力
先にVTKファイルを出力するメンバ関数を作っておきましょう。LocalDensityCalculatorクラスに以下のようなメンバ関数を追加しておきます。
void write_vtk(const std::string &filename, const std::vector<double> &data) {
std::ofstream ofs(filename);
if (!ofs) {
std::cerr << "Error: Could not open file for writing: " << filename << std::endl;
return;
}
ofs << "# vtk DataFile Version 1.0\n";
ofs << "test\n";
ofs << "ASCII\n";
ofs << "DATASET STRUCTURED_POINTS\n";
ofs << "DIMENSIONS " << nx_ << " " << ny_ << " " << nz_ << "\n";
ofs << "ORIGIN 0.0 0.0 0.0\n";
ofs << "SPACING 1.0 1.0 1.0\n";
ofs << "\n";
ofs << "POINT_DATA " << total_cells_ << "\n";
ofs << "\n";
ofs << "SCALARS intensity float\n";
ofs << "LOOKUP_TABLE default\n";
for (double v : data) {
ofs << v << "\n";
}
ofs.close();
std::cerr << filename << std::endl;
}
局所密度の計算
スカラーデータを受け取って、連番のVTKファイルを出力するだけです。この関数にわたすdataを作る関数calc_densityは以下のようになるでしょう。
void calc_density(const std::unique_ptr<lammpstrj::SystemInfo> &si,
const std::vector<lammpstrj::Atom> &atoms) {
std::vector<double> density(total_cells_, 0.0);
for (const auto &atom : atoms) {
double x = atom.x;
double y = atom.y;
double z = atom.z;
if (x < 0.0) x += si->LX;
if (y < 0.0) y += si->LY;
if (z < 0.0) z += si->LZ;
if (x >= si->LX) x -= si->LX;
if (y >= si->LY) y -= si->LY;
if (z >= si->LZ) z -= si->LZ;
int ix = static_cast<int>(x / mx_);
int iy = static_cast<int>(y / my_);
int iz = static_cast<int>(z / mz_);
int index = ix + nx_ * (iy + ny_ * iz);
if (atom.type == 1) {
density[index] += 1.0;
}
}
// 密度に変換(個数密度: 個数 / セル体積)
const double cell_volume = mx_ * my_ * mz_;
for (auto &d : density) {
d /= cell_volume;
}
std::ostringstream oss;
oss << "density." << std::setfill('0') << std::setw(4) << frame_ << ".vtk";
write_vtk(oss.str(), density);
frame_++;
}
特に難しいところはないと思いますが、一点注意すべきは、原子の座標の周期境界条件補正をしている場所です。
if (x < 0.0) x += si->LX;
if (y < 0.0) y += si->LY;
if (z < 0.0) z += si->LZ;
if (x >= si->LX) x -= si->LX;
if (y >= si->LY) y -= si->LY;
if (z >= si->LZ) z -= si->LZ;
LAMMPSはたまにしか周期境界条件補正をしないため、ダンプファイルにはほぼ確実にシミュレーションボックスの外にいる原子が存在します。その座標を修正してやる必要があります。
あとは、先程と同様にこのcalc_densityをコールバック関数としてlammpstrj::for_each_frameに渡してやればOKです。以下のようにラムダ式経由で渡すメンバ関数analyzeを作ります。
void analyze(void){
auto callback_function = [this](const std::unique_ptr<lammpstrj::SystemInfo> &si, const std::vector<lammpstrj::Atom> &atoms){calc_density(si, atoms);};
lammpstrj::for_each_frame(filename_, callback_function);
}
この関数は外から呼ぶのでpublicである必要があります。
これで完成です。
相分離シミュレーション
先ほどのシミュレーションでは短すぎるので、ステップ数を5倍にしましょう。test.inputのrunを25000にします。
run 25000
LAMMPSを実行します。
lmp_serial -i test.input
実行後、出力されたtest.lammptrjをVMDで見るとこんな感じになります。
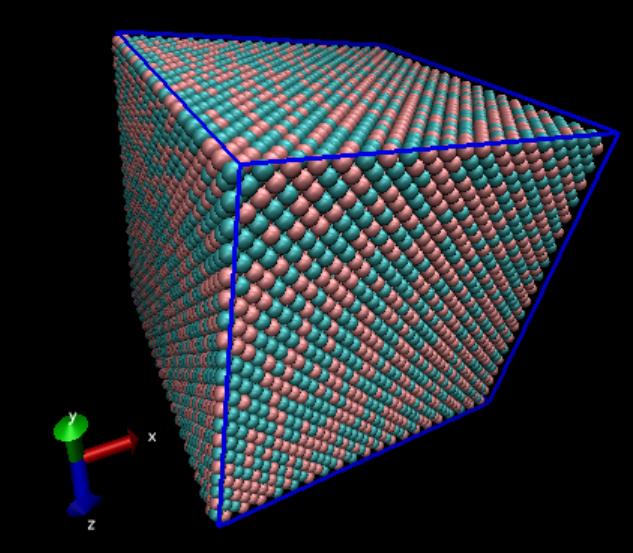
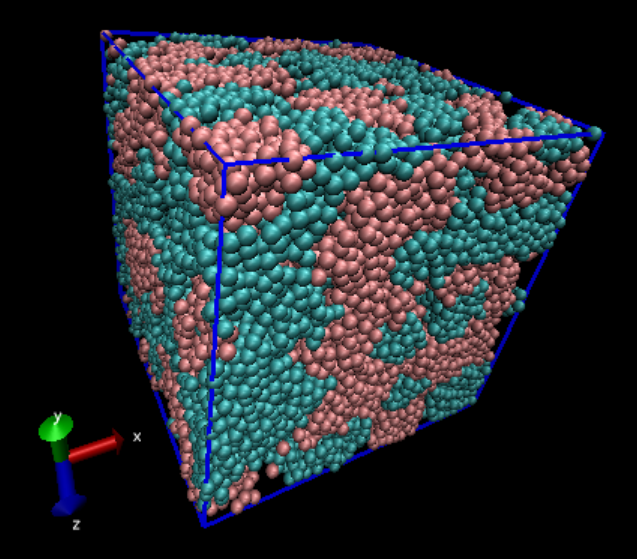
最初に混ざっていた二種類の原子が、終状態では分離していることがわかります。
ではVTKを出力しましょう。ビルドして実行すると以下のような出力がされます。
$ ./lammpstrj-sample
(LX, LY, LZ) = (40.000000, 40.000000, 40.000000)
N = 32000
Requested mesh size: 2
Actual cell size (mx, my, mz): 2, 2, 2
Number of cells (nx, ny, nz): 20, 20, 20
density.0000.vtk
density.0001.vtk
density.0002.vtk
(snip)
density.0046.vtk
density.0047.vtk
density.0048.vtk
density.0049.vtk
density.0050.vtk
出力されたVTKファイルをParaViewで読み込んで適切に可視化するとこんな感じになります。

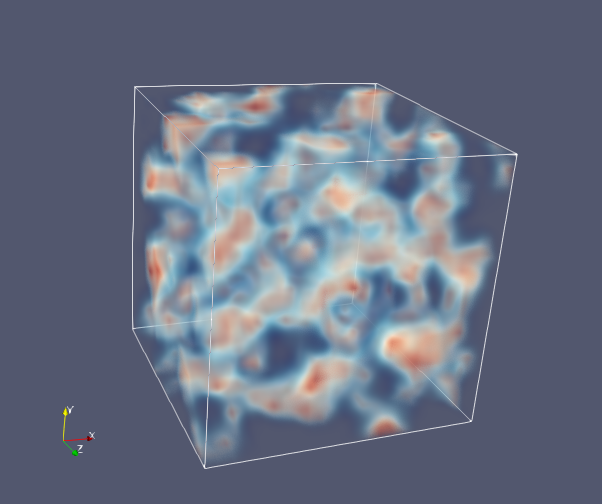
アニメーションGIFにするとこんな感じです。

まとめ
lammpstrjファイルを解析するlammpstrj-parserの使い方を紹介してみました。シングルヘッダーライブラリなのでインクルードするだけで使えます。使う関数は
lammpstrj::read_info(filename)システム情報の取得lammpstrj::for_each_frame(filename_, callback_function)コールバック関数を登録して各フレームごとに処理lammpstrj::for_each_frame(index, filename_, callback_function)コールバック関数を登録して、指定のフレームだけ処理
の3つです。
lammpstrj-parserを使ったコードとして、原子の位置を可視化するtrj-renderも作りました。
lammpstrjの可視化はVMDを使うと思いますが、原子数が多いと時間がかかります。また、スパコンからファイルを持ってくるのも面倒です。trj-renderは、マウスでグリグリはできないものの、その場でPNGファイルに吐いてくれるので便利です。また、指定のフレームだけを可視化できるので、最終フレームだけを確認したりできます。
LAMMPSを使っていて、解析が面倒だと思った方は、是非lammpstrj-parserを使ってみてください。